
Site Map
Latest Release
- Please cite: Langmead B, Schatz MC, Lin J, Pop M, Salzberg SL. Searching for SNPs with cloud computing. Genome Biol 10:R134.
- For release updates, subscribe to the mailing list.
| Crossbow 1.2.1 | 5/30/13 |
|
|
|
|
|
|
Related Tools
| Bowtie: Ultrafast short read alignment |
| Hadoop: Open Source MapReduce |
| Contrail: Cloud-based de novo assembly |
| CloudBurst: Sensitive MapReduce alignment |
| Myrna: Cloud, differential gene expression |
| Tophat: RNA-Seq splice junction mapper |
| Cufflinks: Isoform assembly, quantitation |
| SoapSNP: Accurate SNP/consensus calling |
Reference jars
Related publications
- Langmead B, Schatz M, Lin J, Pop M, Salzberg SL. Searching for SNPs with cloud computing. Genome Biology 10:R134.
- Schatz M, Langmead B, Salzberg SL. Cloud computing and the DNA data race. Nature Biotechnology 2010 Jul;28(7):691-3.
- Langmead B, Hansen K, Leek J. Cloud-scale RNA-sequencing differential expression analysis with Myrna. Genome Biology 11:R83.
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology 10:R25.
- Li R, Li Y, Fang X, Yang H, Wang J, Kristiansen K, Wang J. SNP detection for massively parallel whole-genome resequencing. Genome Res. 2009. 19: 1124-1132.
Authors
Other Documentation
- WABI 09 Poster (.pdf)
Links
Table of Contents
Version 1.2.0
- What is Crossbow?
- A word of caution
- Crossbow modes and prerequisites
- Preparing to run on Amazon Elastic MapReduce
- Installing Crossbow
- Running Crossbow
- Running Crossbow on EMR via the EMR web interface
- Running Crossbow on EMR via the command line
- Running Crossbow on a Hadoop cluster via the command line
- Running Crossbow on a single computer via the command line
- General Crossbow options
- Crossbow examples
- Manifest files
- Reference jars
- Monitoring, debugging and logging
- Crossbow Output
- Other reading
- Acknowledgements
What is Crossbow?
Crossbow is a scalable, portable, and automatic Cloud Computing tool for finding SNPs from short read data. Crossbow employs Bowtie and a modified version of SOAPsnp to perform the short read alignment and SNP calling respectively. Crossbow is designed to be easy to run (a) in "the cloud" (in this case, Amazon's Elastic MapReduce service), (b) on any Hadoop cluster, or (c) on any single computer, without Hadoop. Crossbow exploits the availability of multiple computers and processors where possible.
A word of caution
Renting resources from Amazon Web Services (AKA AWS), costs money, regardless of whether your experiment ultimately succeeds or fails. In some cases, Crossbow or its documentation may be partially to blame for a failed experiment. While we are happy to accept bug reports, we do not accept responsibility for financial damage caused by these errors. Crossbow is provided "as is" with no warranty. See LICENSE file.
Crossbow modes and prerequisites
Crossbow can be run in four different ways.
- Via the Crossbow web interface
In this case, the Crossbow code and the user interface are installed on EC2 web servers. Also, the computers running the Crossbow computation are rented from Amazon, and the user must have EC2, EMR, S3 and SimpleDB accounts and must pay the going rate for the resources used. The user does not need any special software besides a web browser and, in most cases, an S3 tool.
- On Amazon Elastic MapReduce via the command-line
In this case, the Crossbow code is hosted by Amazon and the computers running the Crossbow computation are rented from Amazon. However, the user must install and run (a) the Crossbow scripts, which require Perl 5.6 or later, (b) Amazon's elastic-mapreduce script, which requires Ruby 1.8 or later, and (c) an S3 tool. The user must have EC2, EMR, S3 and SimpleDB accounts and must pay the going rate for the resources used.
- On a Hadoop cluster via the command-line
In this case, the Crossbow code is hosted on your Hadoop cluster, as are supporting tools: Bowtie, SOAPsnp, and possibly [fastq-dump]. Supporting tools must be installed on all cluster nodes, but the Crossbow scripts need only be installed on the master. Crossbow was tested with Hadoop versions 0.20 and 0.20.205, and might also be compatible with other versions newer than 0.20. Crossbow scripts require Perl 5.6 or later.
- On any computer via the command-line
In this case, the Crossbow code and all supporting tools (Bowtie, SOAPsnp, and possibly [fastq-dump]) must be installed on the computer running Crossbow. Crossbow scripts require Perl 5.6 or later. The user specifies the maximum number of CPUs that Crossbow should use at a time. This mode does not require Java or Hadoop.
Preparing to run on Amazon Elastic MapReduce
Before running Crossbow on EMR, you must have an AWS account with the appropriate features enabled. You may also need to install Amazon's elastic-mapreduce tool. In addition, you may want to install an S3 tool, though most users can simply use Amazon's web interface for S3, which requires no installation.
If you plan to run Crossbow exclusively on a single computer or on a Hadoop cluster, you can skip this section.
Create an AWS account by navigating to the AWS page. Click "Sign Up Now" in the upper right-hand corner and follow the instructions. You will be asked to accept the AWS Customer Agreement.
Sign up for EC2 and S3. Navigate to the Amazon EC2 page, click on "Sign Up For Amazon EC2" and follow the instructions. This step requires you to enter credit card information. Once this is complete, your AWS account will be permitted to use EC2 and S3, which are required.
Sign up for EMR. Navigate to the Elastic MapReduce page, click on "Sign up for Elastic MapReduce" and follow the instructions. Once this is complete, your AWS account will be permitted to use EMR, which is required.
Sign up for SimpleDB. With SimpleDB enabled, you have the option of using the AWS Console's Job Flow Debugging feature. This is a convenient way to monitor your job's progress and diagnose errors.
Optional: Request an increase to your instance limit. By default, Amazon allows you to allocate EC2 clusters with up to 20 instances (virtual computers). To be permitted to work with more instances, fill in the form on the Request to Increase page. You may have to speak to an Amazon representative and/or wait several business days before your request is granted.
To see a list of AWS services you've already signed up for, see your Account Activity page. If "Amazon Elastic Compute Cloud", "Amazon Simple Storage Service", "Amazon Elastic MapReduce" and "Amazon SimpleDB" all appear there, you are ready to proceed.
Be sure to make a note of the various numbers and names associated with your accounts, especially your Access Key ID, Secret Access Key, and your EC2 key pair name. You will have to refer to these and other account details in the future.
Installing Amazon's elastic-mapreduce tool
Read this section if you plan to run Crossbow on Elastic MapReduce via the command-line tool. Skip this section if you are not using EMR or if you plan to run exclusively via the Crossbow web interface.
To install Amazon's elastic-mapreduce tool, follow the instructions in Amazon Elastic MapReduce developer's guide for How to Download and Install Ruby and the Command Line Interface. That document describes:
Installing an appropriate version of Ruby, if necessary.
Setting up an EC2 keypair, if necessary.
Setting up a credentials file, which is used by the
elastic-mapreducetool for authentication.
For convenience, we suggest you name the credentials file credentials.json and place it in the same directory with the elastic-mapreduce script. Otherwise you will have to specify the credential file path with the --credentials option each time you run cb_emr.
We strongly recommend using a version of the elastic-mapreduce Ruby script released on or after December 8, 2011. This is when the script switched to using Hadoop v0.20.205 by default, which is the preferred way of running Myrna.
We also recommend that you add the directory containing the elastic-mapreduce tool to your PATH. This allows Crossbow to locate it automatically. Alternately, you can specify the path to the elastic-mapreduce tool via the --emr-script option when running cb_emr.
S3 tools
Running on EMR requires exchanging files via the cloud-based S3 filesystem. S3 is organized as a collection of S3 buckets in a global namespace. S3 charges are incurred when transferring data to and from S3 (but transfers between EC2 and S3 are free), and a per-GB-per-month charge applies when data is stored in S3 over time.
To transfer files to and from S3, use an S3 tool. Amazon's AWS Console has an S3 tab that provides a friendly web-based interface to S3, and doesn't require any software installation. s3cmd is a very good command-line tool that requires Python 2.4 or later. S3Fox Organizer is another GUI tool that works as a Firefox extension. Other tools include Cyberduck (for Mac OS 10.6 or later) and Bucket Explorer (for Mac, Windows or Linux, but commercial software).
Installing Crossbow
Crossbow consists of a set of Perl and shell scripts, plus supporting tools: Bowtie and SOAPsnp . If you plan to run Crossbow via the Crossbow web interface exclusively, there is nothing to install. Otherwise:
Download the desired version of Crossbow from the sourceforge site
Set the
CROSSBOW_HOMEenvironment variable to point to the extracted directory (containingcb_emr)If you plan to run on a local computer or Hadoop cluster:
If using Linux or Mac OS 10.6 or later, you likely don't have to install Bowtie or SOAPsnp, as Crossbow comes with compatible versions of both pre-installed. Test this by running:
$CROSSBOW_HOME/cb_local --testIf the install test passes, installation is complete.
If the install test indicates Bowtie is not installed, obtain or build a
bowtiebinary v0.12.8 or higher and install it by setting theCROSSBOW_BOWTIE_HOMEenvironment variable tobowtie's enclosing directory. Alternately, add the enclosing directory to yourPATHor specify the full path tobowtievia the--bowtieoption when running Crossbow scripts.If the install test indicates that SOAPsnp is not installed, build the
soapsnpbinary using the sources and makefile inCROSSBOW_HOME/soapsnp. You must have compiler tools such as GNUmakeandg++installed for this to work. If you are using a Mac, you may need to install the Apple developer tools. To build thesoapsnpbinary, run:make -C $CROSSBOW_HOME/soapsnpNow install
soapsnpby setting theCROSSBOW_SOAPSNP_HOMEenvironment variable tosoapsnp's enclosing directory. Alternately, add the enclosing directory to yourPATHor specify the full path tosoapsnpvia the--soapsnpoption when running Crossbow scripts.If you plan to run on a Hadoop cluster, you may need to manually copy the
bowtieandsoapsnpexecutables, and possibly also thefastq-dumpexecutable, to the same path on each of your Hadoop cluster nodes. You can avoid this step by installingbowtie,soapsnpandfastq-dumpon a filesystem shared by all Hadoop nodes (e.g. an NFS share). You can also skip this step if Hadoop is installed in pseudo distributed mode, meaning that the cluster really consists of one node whose CPUs are treated as distinct slaves.
The SRA toolkit
The Sequence Read Archive (SRA) is a resource at the National Center for Biotechnology Information (NCBI) for storing sequence data from modern sequencing instruments. Sequence data underlying many studies, including very large studies, can often be downloaded from this archive.
The SRA uses a special file format to store archived read data. These files end in extensions .sra, and they can be specified as inputs to Crossbow's preprocessing step in exactly the same way as FASTQ files.
However, if you plan to use .sra files as input to Crossbow in either Hadoop mode or in single-computer mode, you must first install the SRA toolkit's fastq-dump tool appropriately. See the SRA toolkit page for details about how to download and install.
When searching for the fastq-dump tool at runtime, Crossbow searches the following places in order:
- The path specified in the
--fastq-dumpoption - The directory specified in the
$CROSSBOW_SRATOOLKIT_HOMEenvironment variable. - In the system
PATH
Running Crossbow
The commands for invoking Crossbow from the command line are:
$CROSSBOW_HOME/cb_emr (or just cb_emr if $CROSSBOW_HOME is in the PATH) for running on EMR. See Running Crossbow on EMR via the command line for details.
$CROSSBOW_HOME/cb_hadoop (or just cb_hadoop if $CROSSBOW_HOME is in the PATH) for running on Hadoop. See Running Crossbow on a Hadoop cluster via the command line for details.
$CROSSBOW_HOME/cb_local (or just cb_local if $CROSSBOW_HOME is in the PATH) for running locally on a single computer. See Running Crossbow on a single computer via the command line for details.
Running Crossbow on EMR via the EMR web interface
Prerequisites
- Web browser
- EC2, S3, EMR, and SimpleDB accounts. To check which ones you've already enabled, visit your Account Activity page.
- A tool for browsing and exchanging files with S3
- The AWS Console's S3 tab is a good web-based tool that does not require software installation
- A good command line tool is s3cmd
- A good GUI tool is S3Fox Organizer, which is a Firefox Plugin
- Others include Cyberduck, Bucket Explorer
- Basic knowledge regarding:
- What S3 is, what an S3 bucket is, how to create one, how to upload a file to an S3 bucket from your computer (see your S3 tool's documentation).
- How much AWS resources will cost you
To run
If the input reads have not yet been preprocessed by Crossbow (i.e. input is FASTQ or
.sra), then first (a) prepare a manifest file with URLs pointing to the read files, and (b) upload it to an S3 bucket that you own. See your S3 tool's documentation for how to create a bucket and upload a file to it. The URL for the manifest file will be the input URL for your EMR job.If the input reads have already been preprocessed by Crossbow, make a note of of the S3 URL where they're located. This will be the input URL for your EMR job.
If you are using a pre-built reference jar, make a note of its S3 URL. This will be the reference URL for your EMR job. See the Crossbow website for a list of pre-built reference jars and their URLs.
If you are not using a pre-built reference jar, you may need to build the reference jars and/or upload them to an S3 bucket you own. See your S3 tool's documentation for how to create a bucket and upload to it. The URL for the main reference jar will be the reference URL for your EMR job.
In a web browser, go to the Crossbow web interface.
Fill in the form according to your job's parameters. We recommend filling in and validating the "AWS ID" and "AWS Secret Key" fields first. Also, when entering S3 URLs (e.g. "Input URL" and "Output URL"), we recommend that users validate the entered URLs by clicking the link below it. This avoids failed jobs due to simple URL issues (e.g. non-existence of the "Input URL"). For examples of how to fill in this form, see the E. coli EMR and Mouse chromosome 17 EMR examples.
Running Crossbow on EMR via the command line
Prerequisites
- EC2, S3, EMR, and SimpleDB accounts. To check which ones you've already enabled, visit your Account Activity page.
- A tool for browsing and exchanging files with S3
- The AWS Console's S3 tab is a good web-based tool that does not require software installation
- A good command line tool is s3cmd
- A good GUI tool is S3Fox Organizer, which is a Firefox Plugin
- Others include Cyberduck, Bucket Explorer
- Basic knowledge regarding:
- What S3 is, what an S3 bucket is, how to create one, how to upload a file to an S3 bucket from your computer (see your S3 tool's documentation).
- How much AWS resources will cost you
To run
If the input reads have not yet been preprocessed by Crossbow (i.e. input is FASTQ or
.sra), then first (a) prepare a manifest file with URLs pointing to the read files, and (b) upload it to an S3 bucket that you own. See your S3 tool's documentation for how to create a bucket and upload a file to it. The URL for the manifest file will be the input URL for your EMR job.If the input reads have already been preprocessed by Crossbow, make a note of of the S3 URL where they're located. This will be the input URL for your EMR job.
If you are using a pre-built reference jar, make a note of its S3 URL. This will be the reference URL for your EMR job. See the Crossbow website for a list of pre-built reference jars and their URLs.
If you are not using a pre-built reference jar, you may need to build the reference jars and/or upload them to an S3 bucket you own. See your S3 tool's documentation for how to create a bucket and upload to it. The URL for the main reference jar will be the reference URL for your EMR job.
Run
$CROSSBOW_HOME/cb_emrwith the desired options. Options that are unique to EMR jobs are described in the following section. Options that apply to all running modes are described in the General Crossbow options section. For examples of how to run$CROSSBOW_HOME/cb_emrsee the E. coli EMR and Mouse chromosome 17 EMR examples.
EMR-specific options
|
S3 URL where the reference jar is located. URLs for pre-built reference jars for some commonly studied species (including human and mouse) are available from the Crossbow web site. Note that a Myrna reference jar is not the same as a Crossbow reference jar. If your desired genome and/or SNP annotations are not available in pre-built form, you will have to make your own reference jar and upload it to one of your own S3 buckets (see Reference jars). This option must be specified. |
|
S3 URL where the input is located. If |
|
S3 URL where the output is to be deposited. If |
|
S3 URL where all intermediate results should be be deposited. This can be useful if you later want to resume the computation from partway through the pipeline (e.g. after alignment but before SNP calling). By default, intermediate results are stored in HDFS and disappear once the cluster is terminated. |
|
S3 URL where the preprocessed reads should be stored. This can be useful if you later want to run Crossbow on the same input reads without having to re-run the preprocessing step (i.e. leaving |
|
Local path to the credentials file set up by the user when the |
|
Local path to the |
|
Specify the name by which the job will be identified in the AWS Console. |
|
By default, EMR will terminate the cluster as soon as (a) one of the stages fails, or (b) the job complete successfully. Specify this option to force EMR to keep the cluster alive in either case. |
|
Specify the number of instances (i.e. virtual computers, also called nodes) to be allocated to your cluster. If set to 1, the 1 instance will funcion as both Hadoop master and slave node. If set greater than 1, one instance will function as a Hadoop master and the rest will function as Hadoop slaves. In general, the greater the value of |
|
Specify the type of EC2 instance to use for the computation. See Amazon's list of available instance types and be sure to specify the "API name" of the desired type (e.g. |
|
Pass the specified extra arguments to the |
|
Causes EMR to copy the log files to |
|
By default, Crossbow causes EMR to copy all cluster log files to the |
|
Disables Job Flow Debugging. If this is not specified, you must have a SimpleDB account for Job Flow Debugging to work. You will be subject to additional SimpleDB-related charges if this option is enabled, but those fees are typically small or zero (depending on your account's SimpleDB tier). |
Running Crossbow on a Hadoop cluster via the command line
Prerequisites
Working installation of Hadoop v0.20.2 or v0.20.205. Other versions newer than 0.20 might also work, but haven't been tested.
A
bowtiev0.12.8 executable must exist at the same path on all cluster nodes (including the master). That path must be specified via the--bowtieoption OR located in the directory specified in theCROSSBOW_BOWTIE_HOMEenvironment variable, OR in a subdirectory of$CROSSBOW_HOME/binOR in thePATH(Crossbow looks in that order).$CROSSBOW_HOME/bincomes with pre-built Bowtie binaries for Linux and Mac OS X 10.5 or later. An executable from that directory is used automatically unless the platform is not Mac or Linux or unless overridden by--bowtieor by definingCROSSBOW_BOWTIE_HOME.A Crossbow-customized version of
soapsnpv1.02 must be installed at the same path on all cluster nodes (including the master). That path must be specified via the--soapsnpoption OR located in the directory specified in theCROSSBOW_SOAPSNP_HOMEenvironment variable, OR in a subdirectory of$CROSSBOW_HOME/binOR in thePATH(Crossbow searches in that order).$CROSSBOW_HOME/bincomes with pre-built SOAPsnp binaries for Linux and Mac OS X 10.6 or later. An executable from that directory is used automatically unless the platform is not Mac or Linux or unless overridden by--soapsnpor by definingCROSSBOW_SOAPSNP_HOME.If any of your inputs are in Sequence Read Archive format (i.e. end in
.sra), then thefastq-dumptool from the [SRA Toolkit] must be installed at the same path on all cluster nodes. The path to thefastq-dumptool must be specified via the (--fastq-dump) option ORfastq-dumpmust be located in the directory specified in theCROSSBOW_FASTQ_DUMP_HOMEenvironment variable, ORfastq-dumpmust be found in thePATH(Myrna searches in that order).Sufficient memory must be available on all Hadoop slave nodes to hold the Bowtie index for the desired organism in addition to any other loads placed on those nodes by Hadoop or other programs. For mammalian genomes such as the human genome, this typically means that slave nodes must have at least 5-6 GB of RAM.
To run
Run $CROSSBOW_HOME/cb_hadoop with the desired options. Options that are unique to Hadoop jobs are described in the following subsection. Options that apply to all running modes are described in the General Crossbow options subsection. To see example invocations of $CROSSBOW_HOME/cb_hadoop see the E. coli Hadoop and Mouse chromosome 17 Hadoop examples.
Hadoop-specific options
|
HDFS URL where the reference jar is located. Pre-built reference jars for some commonly studied species (including human and mouse) are available from the Crossbow web site; these can be downloaded and installed in HDFS using |
|
HDFS URL where the input is located. If |
|
HDFS URL where the output is to be deposited. If |
|
HDFS URL where all intermediate results should be be deposited. Default: |
|
HDFS URL where the preprocessed reads should be stored. This can be useful if you later want to run Crossbow on the same input reads without having to re-run the preprocessing step (i.e. leaving |
|
Local path to the Bowtie binary Crossbow should use. |
|
Path to the directory containing |
|
Local path to the SOAPsnp executable to use when running the Call SNPs step. |
Running Crossbow on a single computer via the command line
Prerequisites
A
bowtiev0.12.8 executable must exist on the local computer. The path tobowtiemust be specified via the--bowtieoption OR be located in the directory specified in the$CROSSBOW_BOWTIE_HOMEenvironment variable, OR in a subdirectory of$CROSSBOW_HOME/binOR in thePATH(search proceeds in that order).$CROSSBOW_HOME/bincomes with pre-built Bowtie binaries for Linux and Mac OS X 10.6 or later, so most Mac and Linux users do not need to install either tool.A Crossbow-customized version of
soapsnpv1.02 must exist. The path tosoapsnpmust be specified via the--soapsnpoption OR be in the directory specified in the$CROSSBOW_SOAPSNP_HOMEenvironment variable, OR in a subdirectory of$CROSSBOW_HOME/binOR in thePATH(Crossbow searches in that order).$CROSSBOW_HOME/bincomes with pre-built SOAPsnp binaries for Linux and Mac OS X 10.6 or later. An executable from that directory is used automatically unless the platform is not Mac or Linux or unless overridden by--soapsnpor$CROSSBOW_SOAPSNP_HOME.If any of your inputs are in Sequence Read Archive format (i.e. end in
.sra), then thefastq-dumptool from the [SRA Toolkit] must be installed on the local computer. The path to thefastq-dumptool must be specified via the (--fastq-dump) option ORfastq-dumpmust be located in the directory specified in theMYRNA_FASTQ_DUMP_HOMEenvironment variable, ORfastq-dumpmust be found in thePATH(Myrna searches in that order).Sufficient memory must be available on the local computer to hold one copy of the Bowtie index for the desired organism in addition to all other running workloads. For mammalian genomes such as the human genome, this typically means that the local computer must have at least 5-6 GB of RAM.
To run
Run $CROSSBOW_HOME/cb_local with the desired options. Options unique to local jobs are described in the following subsection. Options that apply to all running modes are described in the General Crossbow options subsection. To see example invocations of $CROSSBOW_HOME/cb_local see the E. coli local and Mouse chromosome 17 local examples.
Local-run-specific options
|
Local path where expanded reference jar is located. Specified path should have a |
|
Local path where the input is located. If |
|
Local path where the output is to be deposited. If |
|
Local path where all intermediate results should be kept temporarily (or permanently, if |
|
Local path where the preprocessed reads should be stored. This can be useful if you later want to run Crossbow on the same input reads without having to re-run the preprocessing step (i.e. leaving |
|
Keep intermediate directories and files, i.e. the output from all stages prior to the final stage. By default these files are deleted as soon as possible. |
|
Keep all temporary files generated during the process of binning and sorting data records and moving them from stage to stage, as well as all intermediate results. By default these files are deleted as soon as possible. |
|
The maximum number of processors to use at any given time during the job. Crossbow will try to make maximal use of the processors allocated. Default: 1. |
|
Maximum number of records to be dispatched to the sort routine at one time when sorting bins before each reduce step. For each child process, this number is effectively divided by the number of CPUs used ( |
|
Maximum number of files that can be opened at once by the sort routine when sorting bins before each reduce step. For each child process, this number is effectively divided by the number of CPUs used ( |
|
Path to the Bowtie executable to use when running the Align step. This overrides all other ways that Crossbow searches for |
|
Path to the directory containing the programs in the SRA toolkit, including |
|
Path to the SOAPsnp executable to use when running the Call SNPs step. This overrides all other ways that Crossbow searches for |
General Crossbow options
The following options can be specified regardless of what mode (EMR, Hadoop or local) Crossbow is run in.
|
Treat all input reads as having the specified quality encoding. |
|
The input path or URL refers to a manifest file rather than a directory of preprocessed reads. The first step in the Crossbow computation will be to preprocess the reads listed in the manifest file and store the preprocessed reads in the intermediate directory or in the |
|
The input path or URL refers to a manifest file rather than a directory of preprocessed reads. Crossbow will preprocess the reads listed in the manifest file and store the preprocessed reads in the |
|
Instead of running the Crossbow pipeline all the way through to the end, run the pipeline up to and including the align stage and store the results in the |
|
Resume the Crossbow pipeline from just after the alignment stage. The |
|
Pass the specified arguments to Bowtie for the Align stage. Default: |
|
Randomly discard a fraction of the input reads. E.g. specify |
|
Randomly discard a fraction of the reference bins prior to SNP calling. E.g. specify |
|
Equivalent to setting |
|
Pass the specified arguments to SOAPsnp in the SNP calling stage. These options are passed to SOAPsnp regardless of whether the reference sequence under consideration is diploid or haploid. Default: |
|
Pass the specified arguments to SOAPsnp in the SNP calling stage. when the reference sequence under consideration is haploid. Default: |
|
Pass the specified arguments to SOAPsnp in the SNP calling stage. when the reference sequence under consideration is diploid. Default: |
|
The specified comma-separated list of chromosome names are to be treated as haploid by SOAPsnp. The rest are treated as diploid. Default: all chromosomes are treated as diploid. |
|
If specified, all chromosomes are treated as haploid by SOAPsnp. |
|
The bin size to use when binning alignments into partitions prior to SNP calling. If load imbalance occurrs in the SNP calling step (some tasks taking far longer than others), try decreasing this. Default: 1,000,000. |
|
Just generate a script containing the commands needed to launch the job, but don't run it. The script's location will be printed so that you may run it later. |
|
Instead of running Crossbow, just search for the supporting tools (Bowtie and SOAPsnp) and report whether and how they were found. If running in Cloud Mode, this just tests whether the |
|
Local directory where temporary files (e.g. dynamically generated scripts) should be deposited. Default: |
Crossbow examples
The following subsections guide you step-by-step through examples included with the Crossbow package. Because reads (and sometimes reference jars) must be obtained over the Internet, running these examples requires an active Internet connection.
E. coli (small)
Data for this example is taken from the study by Parkhomchuk et al.
EMR
Via web interface
Identify an S3 bucket to hold the job's input and output. You may need to create an S3 bucket for this purpose. See your S3 tool's documentation.
Use an S3 tool to upload $CROSSBOW_HOME/example/e_coli/small.manifest to the example/e_coli subdirectory in your bucket. You can do so with this s3cmd command:
s3cmd put $CROSSBOW_HOME/example/e_coli/small.manifest s3://<YOUR-BUCKET>/example/e_coli/
Direct your web browser to the Crossbow web interface and fill in the form as below (substituting for <YOUR-BUCKET>):
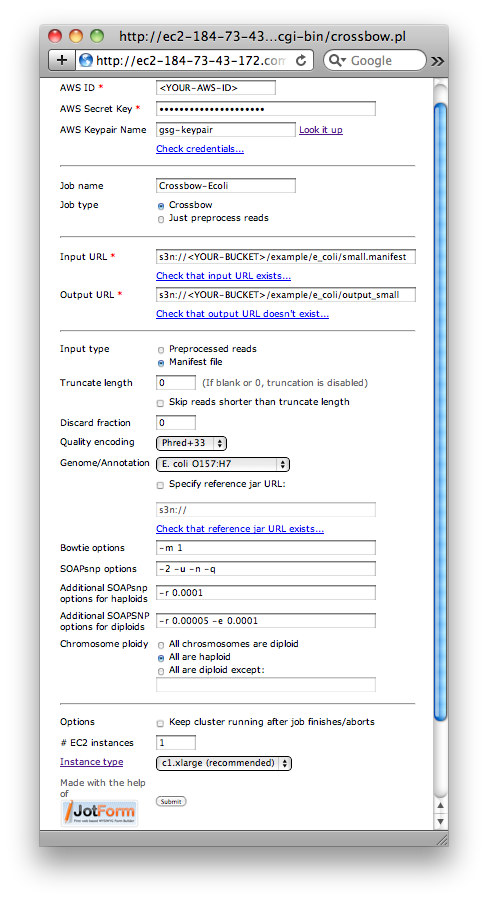
Crossbow web form filled in for the small E. coli example.
- For AWS ID, enter your AWS Access Key ID
- For AWS Secret Key, enter your AWS Secret Access Key
- Optional: For AWS Keypair name, enter the name of your AWS keypair. This is only necessary if you would like to be able to ssh into the EMR cluster while it runs.
- Optional: Check that the AWS ID and Secret Key entered are valid by clicking the "Check credentials..." link
- For Job name, enter
Crossbow-Ecoli - Make sure that Job type is set to "Crossbow"
- For Input URL, enter
s3n://<YOUR-BUCKET>/example/e_coli/small.manifest, substituting for<YOUR-BUCKET> - Optional: Check that the Input URL exists by clicking the "Check that input URL exists..." link
- For Output URL, enter
s3n://<YOUR-BUCKET>/example/e_coli/output_small, substituting for<YOUR-BUCKET> - Optional: Check that the Output URL does not exist by clicking the "Check that output URL doesn't exist..." link
- For Input type, select "Manifest file"
- For Genome/Annotation, select "E. coli" from the drop-down menu
- For Chromosome ploidy, select "All are haploid"
- Click Submit
This job typically takes about 30 minutes on 1 c1.xlarge EC2 node. See Monitoring your EMR jobs for information on how to track job progress. To download the results, use an S3 tool to retrieve the contents of the s3n://<YOUR-BUCKET>/example/e_coli/output_small directory.
Via command line
Test your Crossbow installation by running:
$CROSSBOW_HOME/cb_emr --test
This will warn you if any supporting tools (elastic-mapreduce in this case) cannot be located or run.
Identify an S3 bucket to hold the job's input and output. You may need to create an S3 bucket for this purpose. See your S3 tool's documentation.
Use your S3 tool to upload $CROSSBOW_HOME/example/e_coli/small.manifest to the example/e_coli subdirectory in your bucket. You can do so with this s3cmd command:
s3cmd put $CROSSBOW_HOME/example/e_coli/small.manifest s3://<YOUR-BUCKET>/example/e_coli/
Start the EMR job with the following command (substituting for <YOUR-BUCKET>):
$CROSSBOW_HOME/cb_emr \
--name "Crossbow-Ecoli" \
--preprocess \
--input=s3n://<YOUR-BUCKET>/example/e_coli/small.manifest \
--output=s3n://<YOUR-BUCKET>/example/e_coli/output_small \
--reference=s3n://crossbow-refs/e_coli.jar \
--all-haploids
The --reference option instructs Crossbow to use a pre-built reference jar at URL s3n://crossbow-refs/e_coli.jar. The --preprocess option instructs Crossbow to treat the input as a manifest file, rather than a directory of already-preprocessed reads. As the first stage of the pipeline, Crossbow downloads files specified in the manifest file and preprocesses them into Crossbow's read format. --output specifies where the final output is placed.
This job typically takes about 30 minutes on 1 c1.xlarge EC2 node. See Monitoring your EMR jobs for information on how to track job progress. To download the results, use an S3 tool to retrieve the contents of the s3n://<YOUR-BUCKET>/example/e_coli/output_small directory.
Hadoop
Log into the Hadoop master node and test your Crossbow installation by running:
$CROSSBOW_HOME/cb_hadoop --test
This will tell you if any of the supporting tools or packages are missing on the master. You must also ensure that the same tools are installed in the same paths on all slave nodes, and are runnable by the slaves.
From the master, download the file named e_coli.jar from the following URL:
http://crossbow-refs.s3.amazonaws.com/e_coli.jar
E.g. with this command:
wget http://crossbow-refs.s3.amazonaws.com/e_coli.jar
Equivalently, you can use an S3 tool to download the same file from this URL:
s3n://crossbow-refs/e_coli.jar
E.g. with this s3cmd command:
s3cmd get s3://crossbow-refs/e_coli.jar
Install e_coli.jar in HDFS (the Hadoop distributed filesystem) with the following commands. If the hadoop script is not in your PATH, either add it to your PATH (recommended) or specify the full path to the hadoop script in the following commands.
hadoop dfs -mkdir /crossbow-refs
hadoop dfs -put e_coli.jar /crossbow-refs/e_coli.jar
The first creates a directory in HDFS (you will see a warning message if the directory already exists) and the second copies the local jar files into that directory. In this example, we deposit the jars in the /crossbow-refs directory, but any HDFS directory is fine.
Remove the local e_coli.jar file to save space. E.g.:
rm -f e_coli.jar
Next install the manifest file in HDFS:
hadoop dfs -mkdir /crossbow/example/e_coli
hadoop dfs -put $CROSSBOW_HOME/example/e_coli/small.manifest /crossbow/example/e_coli/small.manifest
Now start the job by running:
$CROSSBOW_HOME/cb_hadoop \
--preprocess \
--input=hdfs:///crossbow/example/e_coli/small.manifest \
--output=hdfs:///crossbow/example/e_coli/output_small \
--reference=hdfs:///crossbow-refs/e_coli.jar \
--all-haploids
The --preprocess option instructs Crossbow to treat the input as a manifest file. As the first stage of the pipeline, Crossbow will download the files specified on each line of the manifest file and preprocess them into Crossbow's read format. The --reference option specifies the location of the reference jar contents. The --output option specifies where the final output is placed.
Single computer
Test your Crossbow installation by running:
$CROSSBOW_HOME/cb_local --test
This will warn you if any supporting tools (bowtie and soapsnp in this case) cannot be located or run.
If you don't already have a CROSSBOW_REFS directory, choose one; it will be the default path Crossbow searches for reference jars. Permanently set the CROSSBOW_REFS environment variable to the selected directory.
Create a subdirectory called $CROSSBOW_REFS/e_coli:
mkdir $CROSSBOW_REFS/e_coli
Download e_coli.jar from the following URL to the new e_coli directory:
http://crossbow-refs.s3.amazonaws.com/e_coli.jar
E.g. with this command:
wget -O $CROSSBOW_REFS/e_coli/e_coli.jar http://crossbow-refs.s3.amazonaws.com/e_coli.jar
Equivalently, you can use an S3 tool to download the same file from this URL:
s3n://crossbow-refs/e_coli.jar
E.g. with this s3cmd command:
s3cmd get s3://crossbow-refs/e_coli.jar $CROSSBOW_REFS/e_coli/e_coli.jar
Change to the new e_coli directory and expand e_coli.jar using an unzip or jar utility:
cd $CROSSBOW_REFS/e_coli && unzip e_coli.jar
Now you may remove e_coli.jar to save space:
rm -f $CROSSBOW_REFS/e_coli/e_coli.jar
Now run Crossbow. Change to the $CROSSBOW_HOME/example/e_coli directory and start the job via the cb_local script:
cd $CROSSBOW_HOME/example/e_coli
$CROSSBOW_HOME/cb_local \
--input=small.manifest \
--preprocess \
--reference=$CROSSBOW_REFS/e_coli \
--output=output_small \
--all-haploids \
--cpus=<CPUS>
Substitute the number of CPUs you'd like to use for <CPUS>.
The --preprocess option instructs Crossbow to treat the input as a manifest file. As the first stage of the pipeline, Crossbow will download the files specified on each line of the manifest file and "preprocess" them into a format understood by Crossbow. The --reference option specifies the location of the reference jar contents. The --output option specifies where the final output is placed. The --cpus option enables Crossbow to use up to the specified number of CPUs at any given time.
Mouse chromosome 17 (large)
Data for this example is taken from the study by Sudbury, Stalker et al.
EMR
Via web interface
First we build a reference jar for a human assembly and annotations using scripts included with Crossbow. The script searches for a bowtie-build executable with the same rules Crossbow uses to search for bowtie. See Installing Crossbow for details. Because one of the steps executed by the script builds an index of the human genome, it should be run on a computer with plenty of memory (at least 4 gigabytes, preferably 6 or more).
cd $CROSSBOW_HOME/reftools
./mm9_chr17_jar
The mm9_chr17_jar script will automatically:
- Download the FASTA sequence for mouse (build mm9) chromome 17 from UCSC.
- Build an index from that FASTA sequence.
- Download the known SNPs and SNP frequencies for mouse chromosome 17 from dbSNP.
- Arrange this information in the directory structure expected by Crossbow.
- Package the information in a jar file named
mm9_chr17.jar.
Next, use an S3 tool to upload the mm9_chr17.jar file to the crossbow-refs subdirectory in your bucket. E.g. with this s3cmd command (substituting for <YOUR-BUCKET>):
s3cmd put $CROSSBOW_HOME/reftools/mm9_chr17/mm9_chr17.jar s3://<YOUR-BUCKET>/crossbow-refs/
You may wish to remove the locally-generated reference jar files to save space. E.g.:
rm -rf $CROSSBOW_HOME/reftools/mm9_chr17
Use an S3 tool to upload $CROSSBOW_HOME/example/mouse17/full.manifest to the example/mouse17 subdirectory in your bucket. E.g. with this s3cmd command:
s3cmd put $CROSSBOW_HOME/example/mouse17/full.manifest s3://<YOUR-BUCKET>/example/mouse17/
Direct your web browser to the Crossbow web interface and fill in the form as below (substituting for <YOUR-BUCKET>):
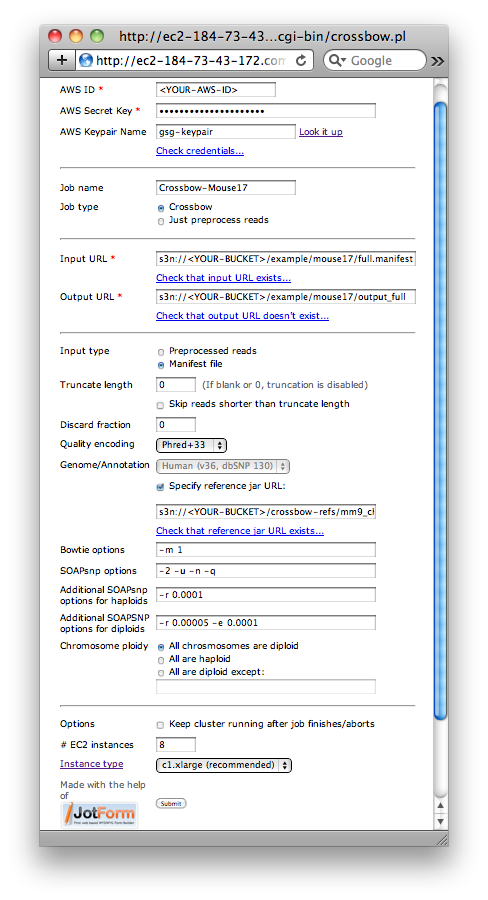
Crossbow web form filled in for the large Mouse Chromosome 17 example.
- For AWS ID, enter your AWS Access Key ID
- For AWS Secret Key, enter your AWS Secret Access Key
- Optional: For AWS Keypair name, enter the name of your AWS keypair. This is only necessary if you would like to be able to ssh into the EMR cluster while it runs.
- Optional: Check that the AWS ID and Secret Key entered are valid by clicking the "Check credentials..." link
- For Job name, enter
Crossbow-Mouse17 - Make sure that Job type is set to "Crossbow"
- For Input URL, enter
s3n://<YOUR-BUCKET>/example/mouse17/full.manifest, substituting for<YOUR-BUCKET> - Optional: Check that the Input URL exists by clicking the "Check that input URL exists..." link
- For Output URL, enter
s3n://<YOUR-BUCKET>/example/mouse17/output_full, substituting for<YOUR-BUCKET> - Optional: Check that the Output URL does not exist by clicking the "Check that output URL doesn't exist..." link
- For Input type, select "Manifest file"
- For Genome/Annotation, check the box labeled "Specify reference jar URL:" and enter
s3n://<YOUR-BUCKET>/crossbow-refs/mm9_chr17.jarin the text box below - Optional: Check that the reference jar URL exists by clicking the "Check that reference jar URL exists..." link
- For Chromosome ploidy, select "All are diploid"
- Click Submit
This job typically takes about 45 minutes on 8 c1.xlarge EC2 instances. See Monitoring your EMR jobs for information on how to track job progress. To download the results, use an S3 tool to retrieve the contents of the s3n://<YOUR-BUCKET>/example/mouse17/output_full directory.
Via command line
First we build a reference jar for a human assembly and annotations using scripts included with Crossbow. The script searches for a bowtie-build executable with the same rules Crossbow uses to search for bowtie. See Installing Crossbow for details. Because one of the steps executed by the script builds an index of the human genome, it should be run on a computer with plenty of memory (at least 4 gigabytes, preferably 6 or more).
cd $CROSSBOW_HOME/reftools
./mm9_chr17_jar
The mm9_chr17_jar script will automatically:
- Download the FASTA sequence for mouse (build mm9) chromome 17 from UCSC.
- Build an index from that FASTA sequence.
- Download the known SNPs and SNP frequencies for mouse chromosome 17 from dbSNP.
- Arrange this information in the directory structure expected by Crossbow.
- Package the information in a jar file named
mm9_chr17.jar.
Next, use an S3 tool to upload the mm9_chr17.jar file to the crossbow-refs subdirectory in your bucket. E.g. with this s3cmd command (substituting for <YOUR-BUCKET>):
s3cmd put $CROSSBOW_HOME/reftools/mm9_chr17/mm9_chr17.jar s3://<YOUR-BUCKET>/crossbow-refs/
You may wish to remove the locally-generated reference jar files to save space. E.g.:
rm -rf $CROSSBOW_HOME/reftools/mm9_chr17
Use an S3 tool to upload $CROSSBOW_HOME/example/mouse17/full.manifest to the example/mouse17 subdirectory in your bucket. E.g. with this s3cmd command:
s3cmd put $CROSSBOW_HOME/example/mouse17/full.manifest s3://<YOUR-BUCKET>/example/mouse17/
To start the EMR job, run the following command (substituting for <YOUR-BUCKET>):
$CROSSBOW_HOME/cb_emr \
--name "Crossbow-Mouse17" \
--preprocess \
--input=s3n://<YOUR-BUCKET>/example/mouse17/full.manifest \
--output=s3n://<YOUR-BUCKET>/example/mouse17/output_full \
--reference=s3n://<YOUR-BUCKET>/crossbow-refs/mm9_chr17.jar \
--instances 8
This job typically takes about 45 minutes on 8 c1.xlarge EC2 instances. See Monitoring your EMR jobs for information on how to track job progress. To download the results, use an S3 tool to retrieve the contents of the s3n://<YOUR-BUCKET>/example/mouse17/output_full directory.
Hadoop
First we build a reference jar for a human assembly and annotations using scripts included with Crossbow. The script searches for a bowtie-build executable with the same rules Crossbow uses to search for bowtie. See Installing Crossbow for details. Because one of the steps executed by the script builds an index of the human genome, it should be run on a computer with plenty of memory (at least 4 gigabytes, preferably 6 or more).
cd $CROSSBOW_HOME/reftools
./mm9_chr17_jar
The mm9_chr17_jar script will automatically:
- Download the FASTA sequence for mouse (build mm9) chromome 17 from UCSC.
- Build an index from that FASTA sequence.
- Download the known SNPs and SNP frequencies for mouse chromosome 17 from dbSNP.
- Arrange this information in the directory structure expected by Crossbow.
- Package the information in a jar file named
mm9_chr17.jar.
Next, use the hadoop script to put the mm9_chr17.jar file in the crossbow-refs HDFS directory. Note tha tif hadoop is not in your PATH, you must specify hadoop's full path instead:
hadoop dfs -mkdir /crossbow-refs
hadoop dfs -put $CROSSBOW_HOME/reftools/mm9_chr17/mm9_chr17.jar /crossbow-refs/mm9_chr17.jar
The first command will yield a warning if the directory already exists; ignore this. In this example, we deposit the jars in the /crossbow-refs directory, but any HDFS directory is fine.
You may wish to remove the locally-generated reference jar files to save space. E.g.:
rm -rf $CROSSBOW_HOME/reftools/mm9_chr17
Now install the manifest file in HDFS:
hadoop dfs -mkdir /crossbow/example/mouse17
hadoop dfs -put $CROSSBOW_HOME/example/mouse17/full.manifest /crossbow/example/mouse17/full.manifest
To start the Hadoop job, run the following command (substituting for <YOUR-BUCKET>):
$CROSSBOW_HOME/cb_hadoop \
--preprocess \
--input=hdfs:///crossbow/example/mouse17/full.manifest \
--output=hdfs:///crossbow/example/mouse17/output_full \
--reference=hdfs:///crossbow-refs/mm9_chr17.jar
Single computer
First we build a reference jar for a human assembly and annotations using scripts included with Crossbow. The script searches for a bowtie-build executable with the same rules Crossbow uses to search for bowtie. See Installing Crossbow for details. Because one of the steps executed by the script builds an index of the human genome, it should be run on a computer with plenty of memory (at least 4 gigabytes, preferably 6 or more).
Run the following commands:
cd $CROSSBOW_HOME/reftools
./mm9_chr17_jar
The mm9_chr17_jar script will automatically:
- Download the FASTA sequence for mouse (build mm9) chromome 17 from UCSC.
- Build an index from that FASTA sequence.
- Download the known SNPs and SNP frequencies for mouse chromosome 17 from dbSNP.
- Arrange this information in the directory structure expected by Crossbow.
- Package the information in a jar file named
mm9_chr17.jar.
Move the directory containing the new reference jar into the $CROSSBOW_REFS directory:
mv $CROSSBOW_HOME/reftools/mm9_chr17 $CROSSBOW_REFS/
Now change to the $CROSSBOW_HOME/example/mouse17 directory and run Crossbow (substitute the number of CPUs you'd like to use for <CPUS>):
cd $CROSSBOW_HOME/example/mouse17
$CROSSBOW_HOME/cb_local \
--input=$CROSSBOW_HOME/example/mouse17/full.manifest \
--preprocess \
--reference=$CROSSBOW_REFS/mm9_chr17 \
--output=output_full \
--cpus=<CPUS>
Manifest files
A manifest file describes a set of FASTQ or .sra formatted input files that might be located:
A manifest file can contain any combination of URLs and local paths from these various types of sources.
FASTQ files can be gzip or bzip2-compressed (i.e. with .gz or .bz2 file extensions). If .sra files are specified in the manifest and Crossbow is being run in single-computer or Hadoop modes, then the fastq-dump tool must be installed and Myrna must be able to locate it. See the --fastq-dump option and the SRA Toolkit section of the manual.
Each line in the manifest file represents either one file, for unpaired input reads, or a pair of files, for paired input reads. For a set of unpaired input reads, the line is formatted:
URL(tab)Optional-MD5
Specifying an MD5 for the input file is optional. If it is specified, Crossbow will attempt to check the integrity of the file after downloading by comparing the observed MD5 to the user-provided MD5. To disable this checking, specify 0 in this field.
For a set of paired input reads, the line is formatted:
URL-1(tab)Optional-MD5-1(tab)URL-2(tab)Optional-MD5-2
Where URL-1 and URL-2 point to input files with all the #1 mates in URL-1 and all the #2 mates in URL-2. The entries in the files must be arranged so that pairs "line up" in parallel. This is commonly the way public paired-end FASTQ datasets, such as those produced by the 1000 Genomes Project, are formatted. Typically these file pairs end in suffixes _1.fastq.gz and _2.fastq.gz.
Manifest files may have comment lines, which must start with the hash (#) symbol, and blank lines. Such lines are ignored by Crossbow.
For examples of manifest files, see the files ending in .manifest in the $CROSSBOW_HOME/example/e_coli and $CROSSBOW_HOME/example/mouse17 directories.
Reference jars
All information about a reference sequence needed by Crossbow is encapsulated in a "reference jar" file. A reference jar includes a set of FASTA files encoding the reference sequences, a Bowtie index of the reference sequence, and a set of files encoding information about known SNPs for the species.
A Crossbow reference jar is organized as:
- A
sequencessubdirectory containing one FASTA file per reference sequence. - An
indexsubdirectory containing the Bowtie index files for the reference sequences. - A
snpssubdirectory containing all of the SNP description files.
The FASTA files in the sequences subdirectory must each be named chrX.fa, where X is the 0-based numeric id of the chromosome or sequence in the file. For example, for a human reference, chromosome 1's FASTA file could be named chr0.fa, chromosome 2 named chr1.fa, etc, all the way up to chromosomes 22, X and Y, named chr21.fa, chr22.fa and chr23.fa. Also, the names of the sequences within the FASTA files must match the number in the file name. I.e., the first line of the FASTA file chr0.fa must be >0.
The index files in the index subdirectory must have the basename index. I.e., the index subdirectory must contain these files:
index.1.ebwt
index.2.ebwt
index.3.ebwt
index.4.ebwt
index.rev.1.ebwt
index.rev.2.ebwt
The index must be built using the bowtie-build tool distributed with Bowtie. When bowtie-build is executed, the FASTA files specified on the command line must be listed in ascending order of numeric id. For instance, for a set of FASTA files encoding human chromosomes 1,2,...,22,X,Y as chr0.fa,chr1.fa,...,chr21.fa, chr22.fa,chr23.fa, the command for bowtie-build must list the FASTA files in that order:
bowtie-build chr0.fa,chr1.fa,...,chr23.fa index
The SNP description files in the snps subdirectory must also have names that match the corresponding FASTA files in the sequences subdirectory, but with extension .snps. E.g. if the sequence file for human Chromosome 1 is named chr0.fa, then the SNP description file for Chromosome 1 must be named chr0.snps. SNP description files may be omitted for some or all chromosomes.
The format of the SNP description files must match the format expected by SOAPsnp's -s option. The format consists of 1 SNP per line, with the following tab-separated fields per SNP:
- Chromosome ID
- 1-based offset into chromosome
- Whether SNP has allele frequency information (1 = yes, 0 = no)
- Whether SNP is validated by experiment (1 = yes, 0 = no)
- Whether SNP is actually an indel (1 = yes, 0 = no)
- Frequency of A allele, as a decimal number
- Frequency of C allele, as a decimal number
- Frequency of T allele, as a decimal number
- Frequency of G allele, as a decimal number
- SNP id (e.g. a dbSNP id such as
rs9976767)
Once these three subdirectories have been created and populated, they can be combined into a single jar file with a command like this:
jar cf ref-XXX.jar sequences snps index
To use ref-XXX.jar with Crossbow, you must copy it to a location where it can be downloaded over the internet via HTTP, FTP, or S3. Once it is placed in such a location, make a note if its URL.
Building a reference jar using automatic scripts
The reftools subdirectory of the Crossbow package contains scripts that assist in building reference jars, including scripts that handle the entire process of building reference jars for hg18 (UCSC human genome build 18) and mm9 (UCSC mouse genome build 9). The db2ssnp script combines SNP and allele frequency information from dbSNP to create a chrX.snps file for the snps subdirectory of the reference jar. The db2ssnp_* scripts drive the db2ssnp script for each chromosome in the hg18 and mm9 genomes. The *_jar scripts drive the entire reference-jar building process, including downloading reference FASTA files, building a Bowtie index, and using db2ssnp to generate the .snp files for hg18 and mm9.
Monitoring, debugging and logging
Single computer
Single-computer runs of Crossbow are relatively easy to monitor and debug. Progress messages are printed to the console as the job runs. When there is a fatal error, Crossbow usually indicates exactly which log file on the local filesystem contains the relevant error message. Additional debugging is possible when intermediate and temporary files are kept rather than discarded; see --keep-intermediates and --keep-all. All output and logs are stored on the local filesystem; see --intermediate and --output options.
Hadoop
The simplest way to monitor Crossbow Hadoop jobs is via the Hadoop JobTracker. The JobTracker is a web server that provides a point-and-click interface for monitoring jobs and reading output and other log files generated by those jobs, including after they've finished.
When a job fails, you can often find the relevant error message by "drilling down" from the "step" level through the "job" level and "task" levels, and finally to the "attempt" level. To diagnose why an attempt failed, click through to the "stderr" ("standard error") log and scan for the relevant error message.
See your version of Hadoop's documentation for details on how to use the web interface. Amazon has a brief document describing How to Use the Hadoop User Interface, though some of the instructions are specific to clusters rented from Amazon. Hadoop, the Definitive Guide is also an excellent reference.
EMR
The recommended way to monitor EMR Hadoop jobs is via the AWS Console. The AWS Console allows you to see:
- The status for job (e.g. "COMPLETED", "RUNNING" or "FAILED")
- The status for each step of each job
- How long a job has been running for and how many "compute units" have been utilized so far.
- The exact Hadoop commands used to initiate each job step.
- The button for Debugging Job Flows
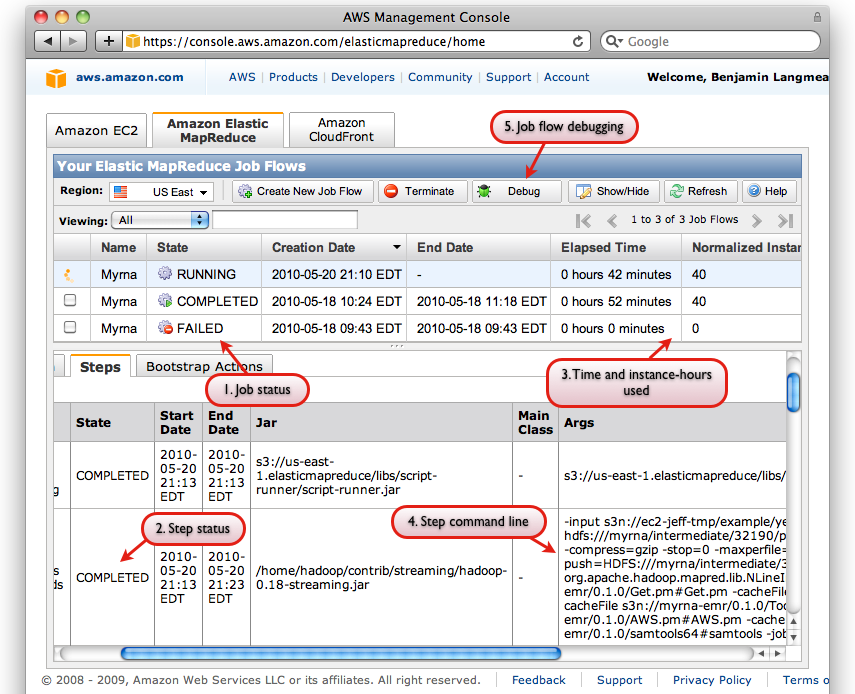
Screen shot of AWS Console interface with some relevant interface elements labeled
The AWS Console also has a useful facility for Debugging Job Flows, which is accessible via the "Debug" button on the "Elastic MapReduce" tab of the Console (labeled "5"). You must (a) have a SimpleDB account (b) not have specified --no-emr-debug in order to use all of the EMR Debug interface's features:
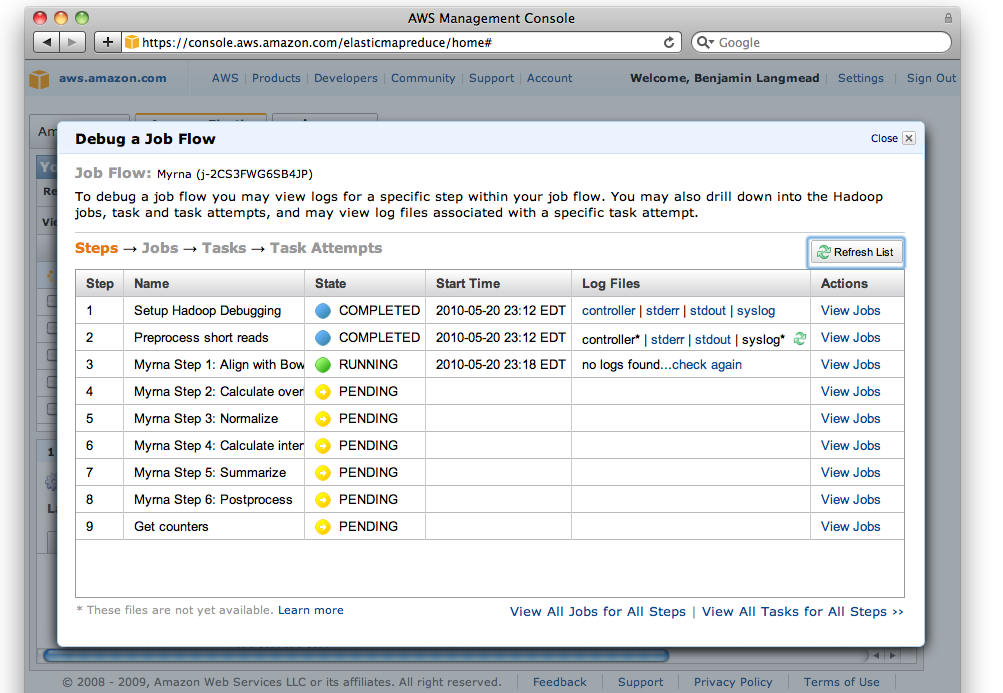
Screen shot of EMR Debug interface
The debug interface is similar to Hadoop's JobTracker interface. When a job fails, you can often find the relevant error message by "drilling down" from the "job" level, through the "task" level, and finally to the "attempt" level. To diagnose why an attempt failed, click through to the "stderr" ("standard error") log and scan for the relevant error message.
For more information, see Amazon's document on Debugging Job Flows.
AWS Management Console
A simple way to monitor your EMR activity is via the AWS Console. The AWS Console summarizes current information regarding all your running EC2 nodes and EMR jobs. Each job is listed in the "Amazon Elastic MapReduce" tab of the console, whereas individual EC2 nodes are listed in the "Amazon EC2" tab.
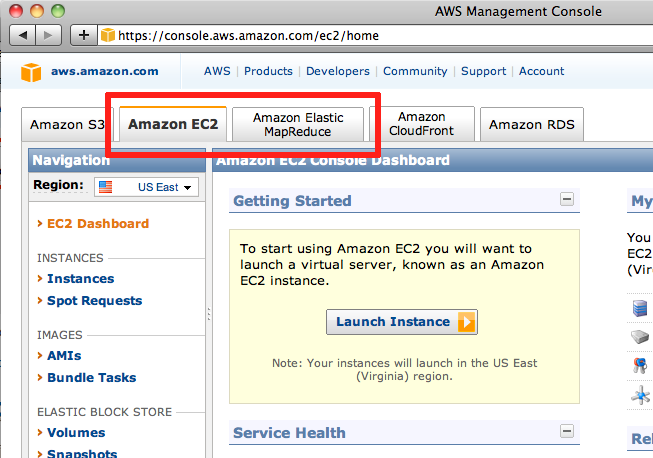
Screen shot of AWS console; note tabs for "Amazon Elastic MapReduce" and "Amazon EC2"
Crossbow Output
Once a Crossbow job completes successfully, the output is deposited in a crossbow_results subdirectory of the specified --output directory or URL. Within the crossbow_results subdirectory, results are organized as one gzipped result file per chromosome. E.g. if your run was against the hg18 build of the human genome, the output files from your experiment will named:
<output_url>/crossbow_results/chr1.gz
<output_url>/crossbow_results/chr2.gz
<output_url>/crossbow_results/chr3.gz
...
<output_url>/crossbow_results/chr21.gz
<output_url>/crossbow_results/chr22.gz
<output_url>/crossbow_results/chrX.gz
<output_url>/crossbow_results/chrY.gz
<output_url>/crossbow_results/chrM.gz
Each individual record is in the SOAPsnp output format. SOAPsnp's format consists of 1 SNP per line with several tab-separated fields per SNP. The fields are:
- Chromosome ID
- 1-based offset into chromosome
- Reference genotype
- Subject genotype
- Quality score of subject genotype
- Best base
- Average quality score of best base
- Count of uniquely aligned reads corroborating the best base
- Count of all aligned reads corroborating the best base
- Second best base
- Average quality score of second best base
- Count of uniquely aligned reads corroborating second best base
- Count of all aligned reads corroborating second best base
- Overall sequencing depth at the site
- Sequencing depth of just the paired alignments at the site
- Rank sum test P-value
- Average copy number of nearby region
- Whether the site is a known SNP from the file specified with
-s
Note that field 15 was added in Crossbow and is not output by unmodified SOAPsnp.
For further details, see the SOAPsnp manual.
Other reading
The Crossbow paper discusses the broad design philosophy of both Crossbow and Myrna and why cloud computing can be considered a useful trend for comparative genomics applications. The Bowtie paper discusses the alignment algorithm underlying Bowtie.
For additional information regarding Amazon EC2, S3, EMR, and related services, see Amazon's AWS Documentation. Some helpful screencasts are posted on the AWS Console home page.
For additional information regarding Hadoop, see the Hadoop web site and Cloudera's Getting Started with Hadoop document. Cloudera's training virtual machine for VMWare is an excellent way to get acquainted with Hadoop without having to install it on a production cluster.
Acknowledgements
Crossbow software is by Ben Langmead and Michael C. Schatz.
Bowtie software is by Ben Langmead and Cole Trapnell.
SOAPsnp is by Ruiqiang Li, Yingrui Li, Xiaodong Fang, Huanming Yang, Jian Wang, Karsten Kristiansen, and Jun Wang.
